![]()
Based on the RNA-Seq workshop by Melbourne Bioinformatics written by Mahtab Mirmomeni, Andrew Lonie, Jessica Chung Original
Modified by David Powell and Stuart Archer
web : platforms.monash.edu/bioinformatics
twitter: [@MonashBioinfo](https://twitter.com/MonashBioinfo)
email: bioinformatics.platform@monash.edu
This tutorial will cover the basics of RNA-seq using Galaxy V4.0.0 and Degust.
It is recommended you have some familiarity with Galaxy, see: Galaxy
The data for this tutorial is from the paper, A comprehensive comparison of RNA-Seq-based transcriptome analysis from reads to differential gene expression and cross-comparison with microarrays: a case study in Saccharomyces cerevisiae by Nookaew et al. [1] which studies S.cerevisiae strain CEN.PK 113-7D (yeast) under two different metabolic conditions: glucose-excess (batch) or glucose-limited (chemostat).
The RNA-Seq data has been uploaded in NCBI, short read archive (SRA), with accession SRS307298. There are 6 samples in total-- two treatments with three biological replicates each sequenced paired-end. We have selected only the first read, and only two replicates of each condition to keep the data small for this workshop.
We have extracted chromosome I reads from the samples to make the tutorial a suitable length. This has implications, as discussed in section 7.
Open a browser and go to the Melbourne Galaxy server : Galaxy-mel.genome.edu.au
If you already have an account, login with your credentials. If not, register as a new user using the 'you may create one' link at top.
You can import the data by:
Upload the sequence data by pasting the following links into the text input area. These two files are single-end samples from the batch condition (glucose-excess). Make sure the type is specified as 'fastqsanger' when uploading (NOT 'fastqcsanger').
https://swift.rc.nectar.org.au:8888/v1/AUTH_a3929895f9e94089ad042c9900e1ee82/RNAseqDGE_ADVNCD/batch1_chrI_1.fastq
https://swift.rc.nectar.org.au:8888/v1/AUTH_a3929895f9e94089ad042c9900e1ee82/RNAseqDGE_ADVNCD/batch2_chrI_1.fastq
These two files are single-end samples from the chem condition (glucose-limited). Make sure the type is specified as 'fastqsanger' when uploading (NOT 'fastqcsanger').
https://swift.rc.nectar.org.au:8888/v1/AUTH_a3929895f9e94089ad042c9900e1ee82/RNAseqDGE_ADVNCD/chem1_chrI_1.fastq
https://swift.rc.nectar.org.au:8888/v1/AUTH_a3929895f9e94089ad042c9900e1ee82/RNAseqDGE_ADVNCD/chem2_chrI_1.fastq
https://swift.rc.nectar.org.au:8888/v1/AUTH_a3929895f9e94089ad042c9900e1ee82/RNAseqDGE_ADVNCD/genes.gtf
batch1_chrI_1.fastqbatch2_chrI_1.fastqchem1_chrI_1.fastqchem2_chrI_1.fastqgenes.gtfNote: Low quality reads have already been trimmed.
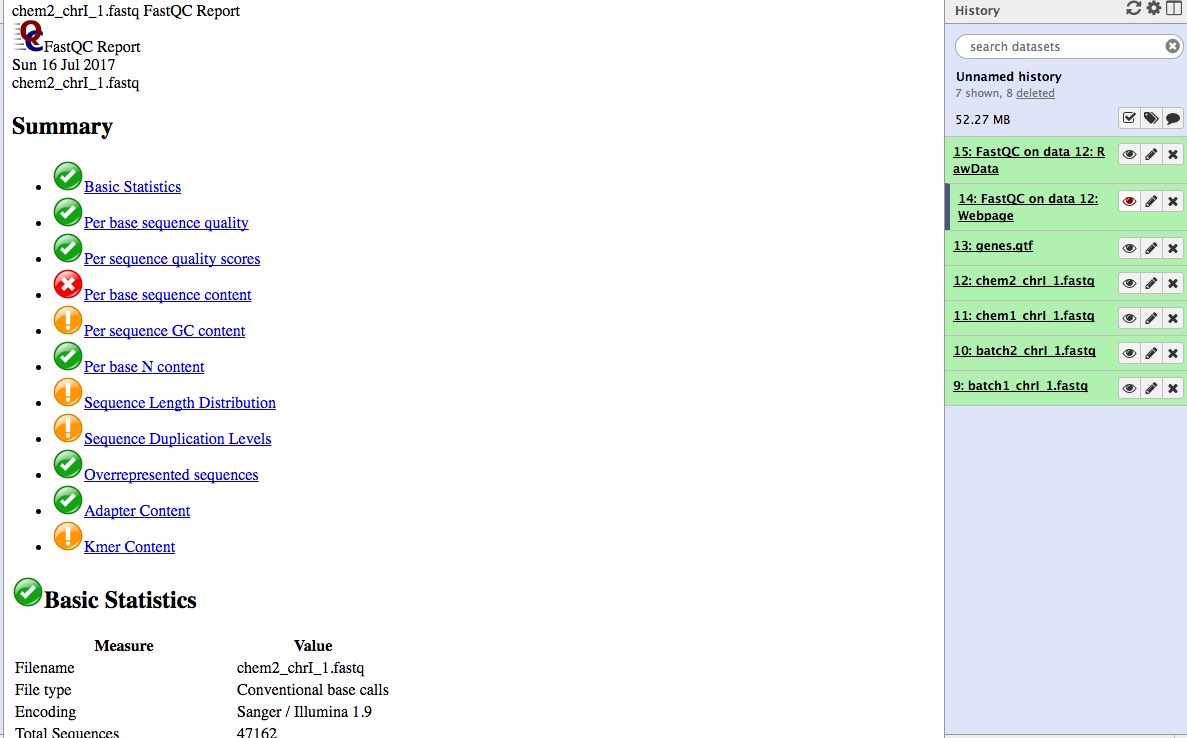
Look at the generated FastQC metrics. This data looks pretty good - high per-base quality scores (most above 30).
In this section we map the reads in our FASTQ files to a reference genome. As these reads originate from mRNA, we expect some of them will cross exon/intron boundaries when we align them to the reference genome. Tophat is a splice-aware mapper for RNA-seq reads that is based on Bowtie. It uses the mapping results from Bowtie to identify splice junctions between exons. More information on Tophat can be found here.
In the left tool panel menu, under NGS ANALYSIS, select NGS: RNA Analysis > Tophat and set the parameters as follows:
batch1_chrI_1.fastqbatch2_chrI_1.fastqchem1_chrI_1.fastqchem2_chrI_1.fastq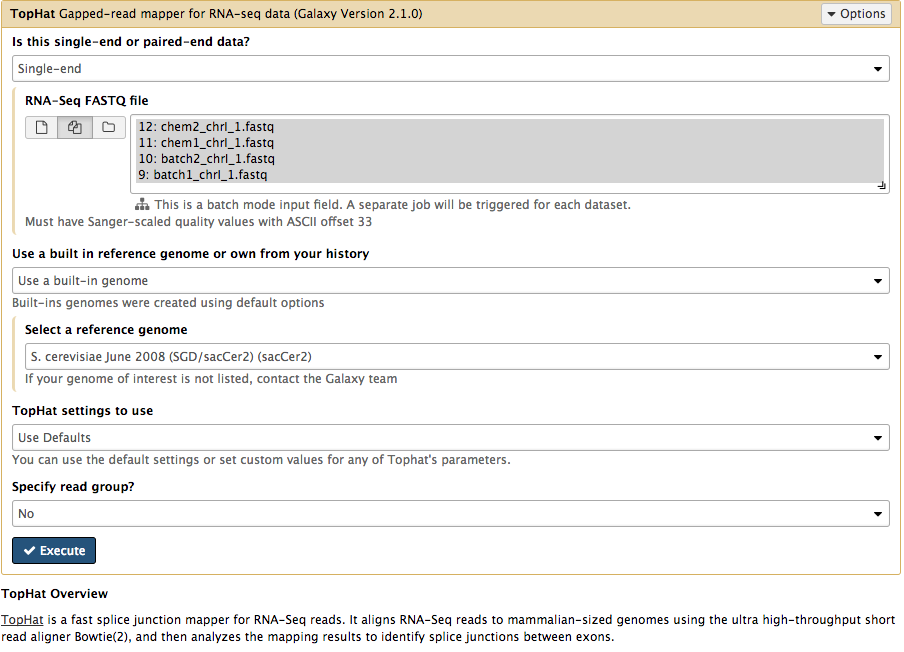
Note: This may take a few minutes, depending on how busy the server is.
You should have 5 output files for each of the FASTQ input files:
You should have a total of 20 Tophat output files in your history.
Rename the 4 accepted_hits files into a more meaningful name (e.g. 'Tophat on data 1: accepted_hits' to 'batch1-accepted_hits.bam') by using the pen icon next to the file.
genes.gtf' file and Execute. A new GFF file should appear, e.g. 'GTF-to-GFF on data 5: Converted'Use a built-in genome' and under Select a reference genome select 'S. cerevisiae June2008 (SGD / sacCer2) (sacCer2)'GFF.GFF3/BED/GBK Features' and choose the GFF file generated in step 3.1 above, e.g. 'GTF-to-GFF on data 5: Converted' Note: leave all other options as default values.BAM Pileups'. Choose a BAM file, e.g. 'batch1-accepted_hits.bam'. Note: leave all other options as default values.Error reading from name store' error, this won't affect your session so just click it away.chrI:86985-87795' to view an intron junction on gene YAL030W.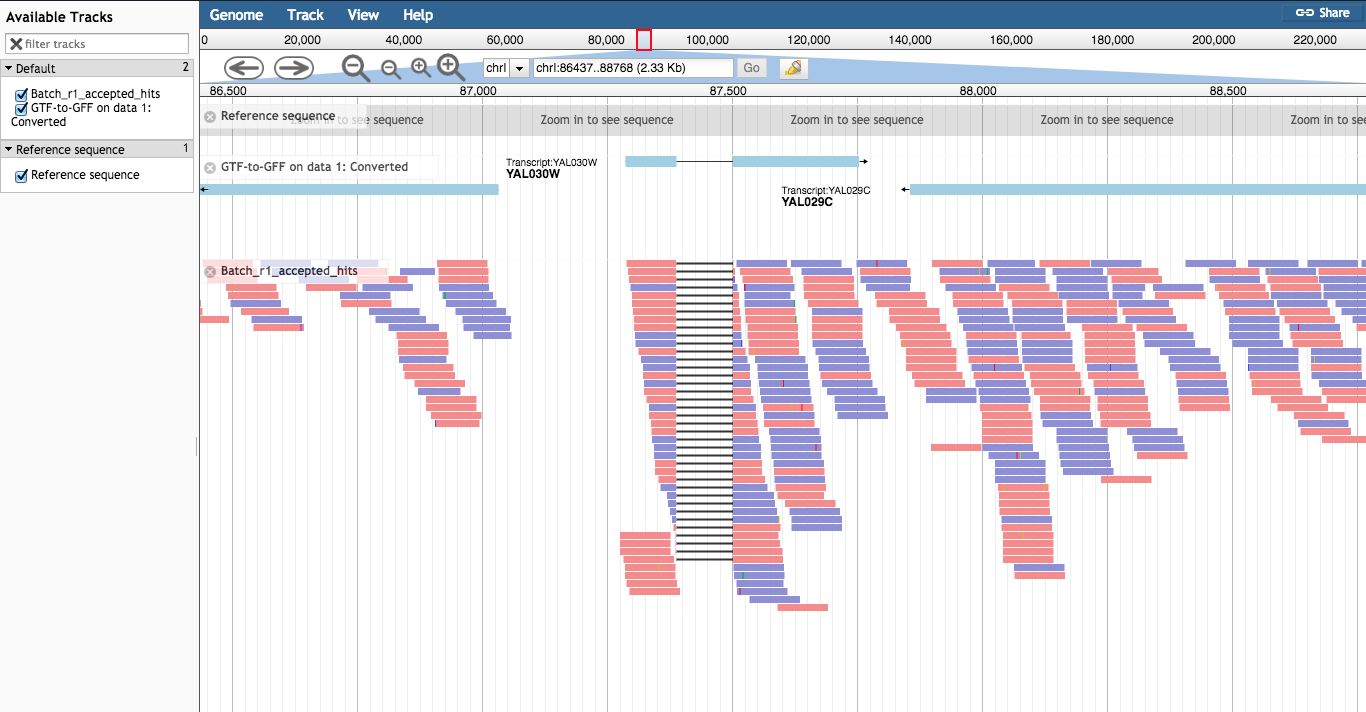
It is useful to understand the BAM/SAM format. Convert one of your BAM files to SAM format, and view the text within Galaxy.
Note: If using Galaxy v4.2.0, BAM files can be viewed directly with the eye icon without conversion to SAM.
featureCounts creates a count matrix using the number of the reads from each bam file that map to the genomic features in the genes.gtf. For each feature (a gene for example) a count matrix shows how many reads were mapped to this feature.
In the left tool panel menu, under NGS ANALYSIS, select NGS: RNA Analysis > SAM/BAM to count matrix and set the parameters as follows:
genes.gtf'batch1-accepted_hits.bambatch2-accepted_hits.bamchem1-accepted_hits.bamchem2-accepted_hits.bamEach column corresponds to a sample and each row corresponds to a gene. By sight, see if you can find a gene you think is differentially expressed from looking at the counts.
We now have a count matrix, with a count against each corresponding sample. We will use this matrix in later sections to calculate the differentially expressed genes.
edgeR is an R package, that is used for analysing differential expression of RNA-Seq data and can either use exact statistical methods or generalised linear models.
In the Galaxy tool panel, under NGS ANALYSIS, select NGS: RNA Analysis > Differential_Count and set the parameters as follows:
bams to DGE count matrix_htseqsams2mx.xls'Differential_Counts_edgeR'Batch'batch1-accepted_hits.bambatch2-accepted_hits.bamChem'chem1-accepted_hits.bamchem2-accepted_hits.bamDifferential_Counts_edgeR_topTable_edgeR.xls' file by clicking on the eye icon. This file is a list of genes sorted by p-value from using EdgeR to perform differential expression analysis.Differential_Counts_edgeR.html' file. This file has some output logs and plots from running edgeR. If you are familiar with R, you can examine the R code used for analysis by scrolling to the bottom of the file, and clicking 'Differential_Counts.Rscript' to download the Rscript file.Under BASIC TOOLS, click on Filter and Sort > Filter:
Differential_Counts_edgeR_topTable_edgeR.xls'This will keep the genes that have an 'adj.p.value' (column 6 in the table) of less or equal to 0.05. This column is the False Discovery Rate estimate if edgeR was run using default options. There should be 47 genes in this file (of which the 5% are possible false discoveries, as estimated by EdgeR). Rename this file by clicking on the pencil icon of and change the name from 'Filter on data x' to 'edgeR_Significant_DE_Genes'.
Degust is an interactive visualiser for analysing RNA-seq data. It runs as a web service and can be found at http://degust.erc.monash.edu/.
bams to DGE count matrix_htseqsams2mx.xls' generated in Section 4 using the disk icon. You can also download this count matrix from here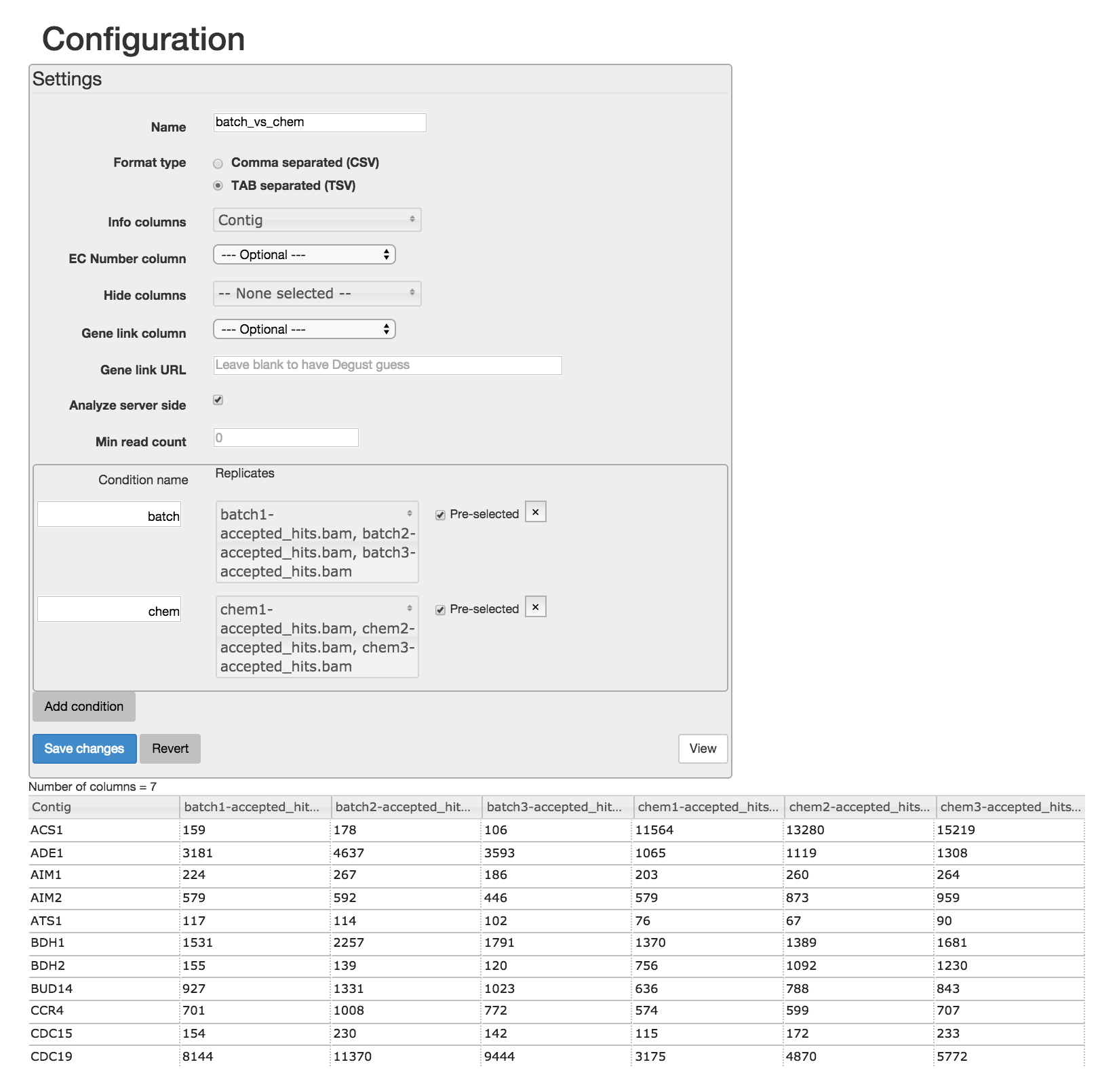
Read through the Degust tour of features. Explore the parallel coordinates plot, MA plot, MDS plot, heatmap and gene list. Each is fully interactive and influences other portions on the display depending on what is selected.
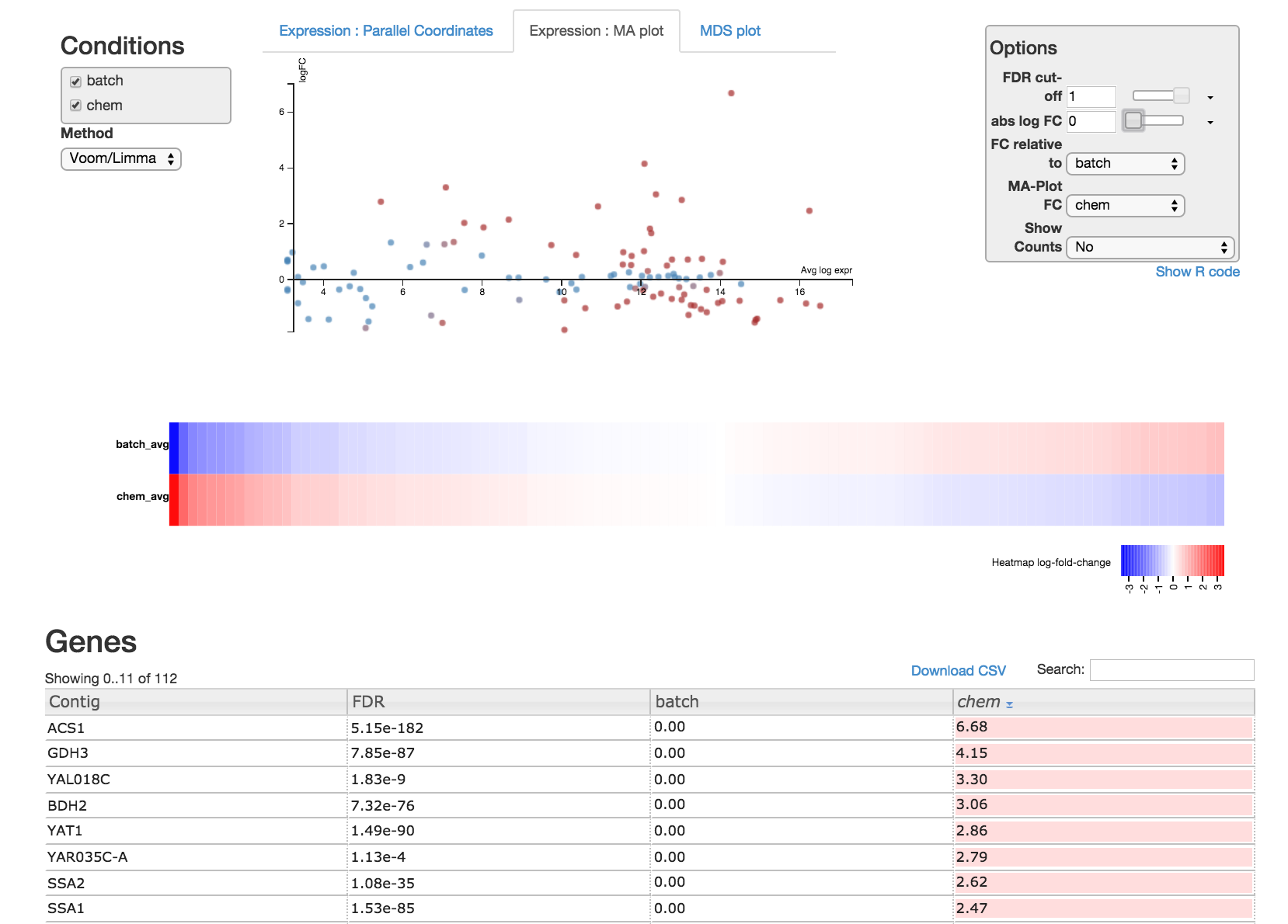
On the right side of the page is an options module which can set thresholds to filter genes using statistical significance or absolute-fold-change.
On the left side is a dropdown box you can specify the method (Voom/Limma or edgeR) used to perform differential expression analysis on the data. You can also view the R code by clicking "Show R code" under the options module on the right.
Degust also provides an example dataset with 4 conditions and more genes. You can play with the demo dataset by clicking on the "Try the demo" button on the Degust homepage. The demo dataset includes a column with an EC number for each gene. This means genes can be displayed on Kegg pathways using the module on the right.
The previous example was a straightforward comparison of two conditions. Here we will look at an experiment with more conditions and consider how to ask interesting questions of the experimental data. We will use the RNA-seq data from this paper : PUMA promotes apoptosis of hematopoietic progenitors driving leukemic progression in a mouse model of myelodysplasia by Guirgas et al. [2] which uses a mouse-model to study Myelodysplastic syndrome. This uses a NUP98-HOXD13 (NHD13) transgenic mouse model, and a mouse model over-expression BCL2 which blocked apoptosis. There are four mouse genotypes used in this experiement - wildtype (WT), BCL2 mutant (B), NHD13 mutant (N), combined mutant (NB).
Download count matrix nhd13.csv. And configure these four conditions, each with 3 replicates. Also, set 'Min gene CPM' to 1, and 'in at least samples' to 3.
Things to do:
abs log FC' and/or 'FDR cut-off', what is driving the separation? Is this a concern?The biological question being asked in the original paper is essentially:
"What is the global response of the yeast transcriptome in the shift from growth at glucose excess conditions (batch) to glucose-limited conditions (chemostat)?"
We can address this question by attempting to interpret our differentially expressed gene list at a higher level, perhaps by examining the categories of gene and protein networks that change in response to glucose.
For example, we can input our list of differentially expressed genes to a Gene Ontology (GO) enrichment analysis tool such as GOrilla to find out the GO enriched terms.
NOTE: Because of time-constraints in this tutorial, the analysis was confined to a single chromosome (chromosome I) and as a consequence we don’t have sufficient information to look for groups of differentially expressed genes (simply because we don’t have enough genes identified from the one chromosome to look for statistically convincing over-representation of any particular gene group).
Download the list of genes here in a plain-text file to your local computer by right clicking on the link and selecting Save Link As...
Note that there are ~2500 significantly differentially expressed genes identified in the full analysis. Also note that the genes are ranked in order of statistical significance. This is critical for the next step.
Once the analysis has finished running, you will be redirected to a page depicting the GO enriched biological processes and its significance (indicated by colour), based on the genes you listed. 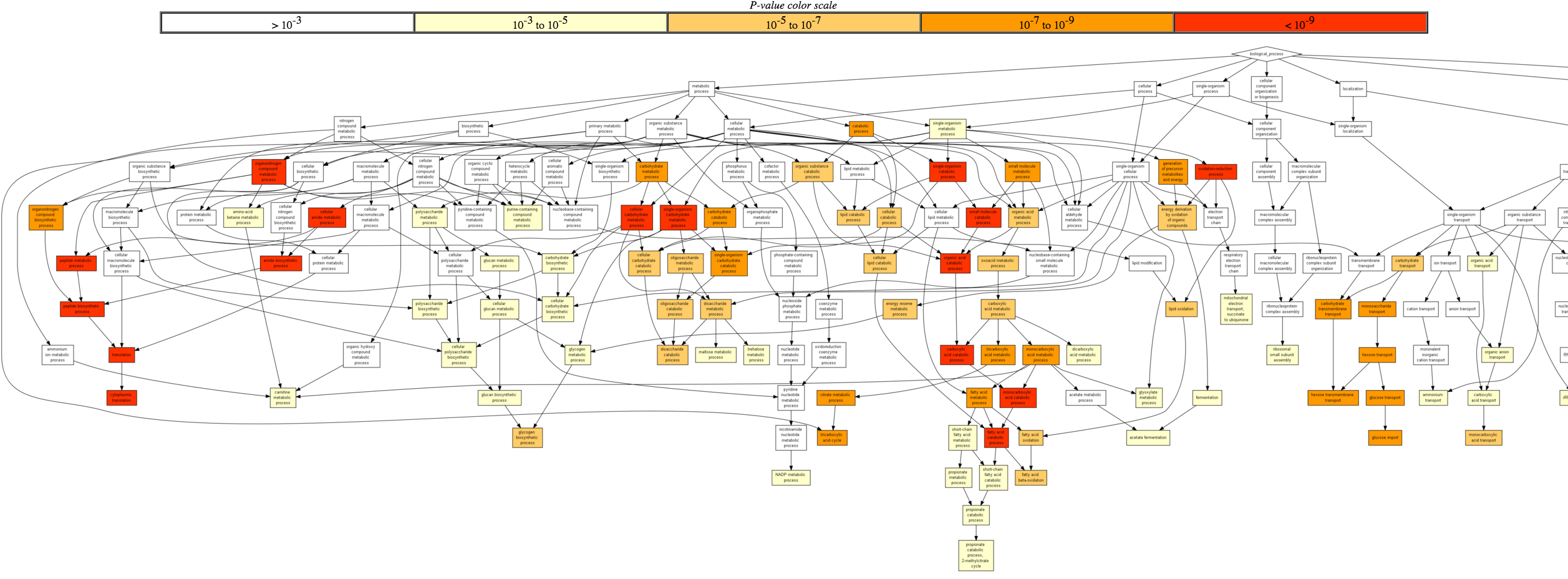
Scroll down to view a table of GO terms and their significance scores. In the last column, you can toggle the [+] Show genes to see the list of associated genes.
Experiment with different ontology categories (Function, Component) in GOrilla.
At this stage you are interpreting the experiment in different ways, potentially discovering information that will lead you to further lab experiments. This is driven by your biological knowledge of the problem space. There are an unlimited number of methods for further interpretation of which GSEA is just one.
The aim in this section is to statistically test for differential expression using Cuffdiff and obtain a list of significant genes.
In the left tool panel menu, under NGS Analysis, select NGS: RNA Analysis > Cuffdiff and set the parameters as follows:
batch1-accepted_hits.bambatch2-accepted_hits.bam (Multiple datasets can be selected by holding down the shift key or the ctrl key (Windows) or the command key (OSX).)chem1-accepted_hits.bamchem2-accepted_hits.bam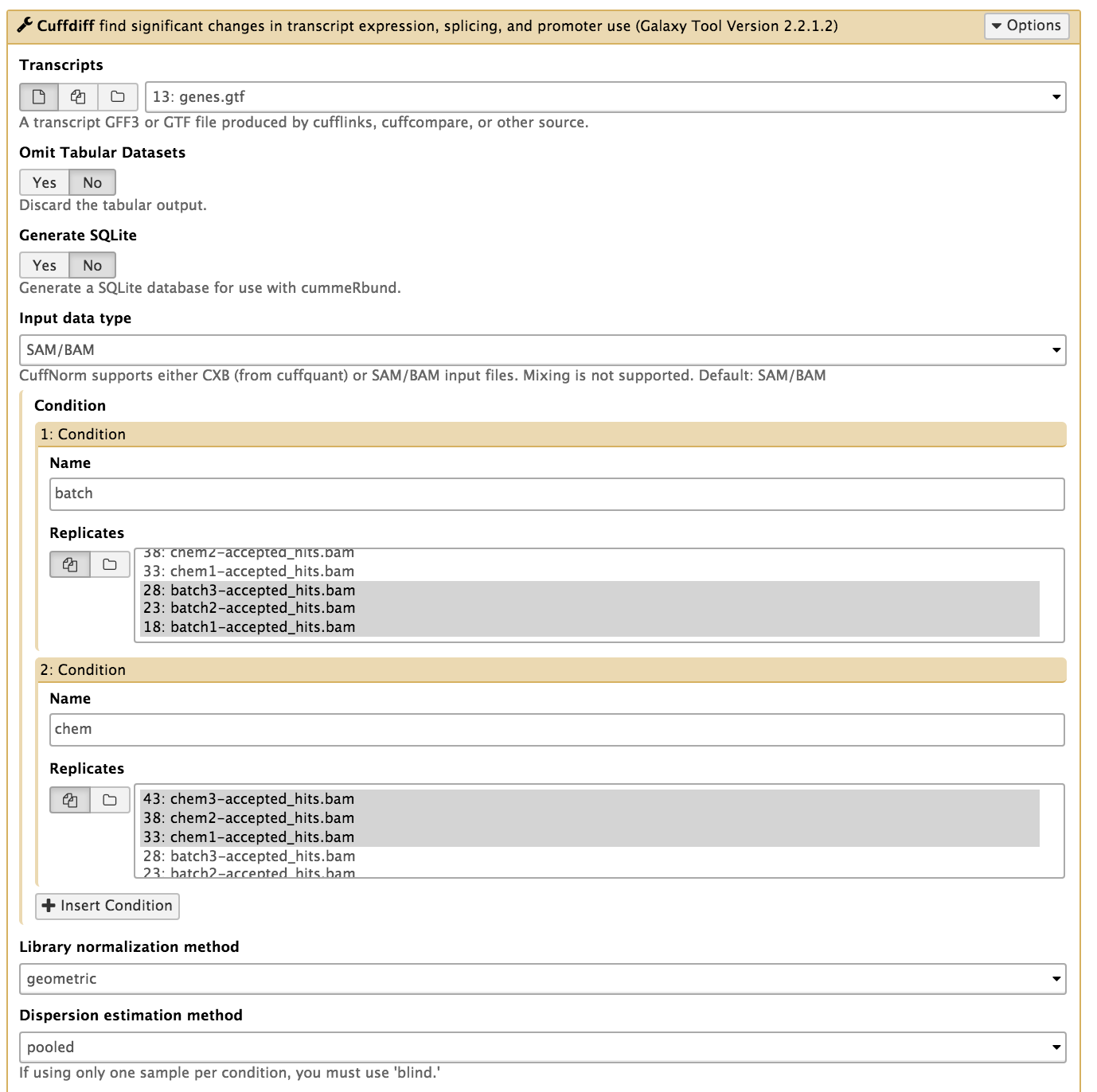
Note: This step may take a while, depending on how busy the server is.
There should be 11 output files from Cuffdiff. These files should all begin with something like "Cuffdiff on data 43, data 38, and others". We will mostly be interested in the file ending with "gene differential expression testing" which contains the statistical results from testing the level of gene expression between the batch condition and chem condition.
Filter based on column 14 ("significant") - a binary assessment of q_value > 0.05, where q_value is p_value adjusted for multiple testing. Under Basic Tools, click on Filter and Sort > Filter:
This will keep only those entries that Cuffdiff has marked as significantly differentially expressed. There should be 53 differentially expressed genes in this list.
We can rename this file by clicking on the pencil icon of the outputted file and change the name from "Filter on data x" to Cuffdiff_Significant_DE_Genes.
DESeq2 is an R package that uses a negative binomial statistical model to find differentially expressed genes. It can work without replicates (unlike edgeR) but the author strongly advises against this for reasons of statistical validity.
In the Galaxy tool panel, under NGS Analysis, select NGS: RNA Analysis > Differential_Count and set the parameters as follows:
matrix_htseqsams2mx.xlsbatch1-accepted_hits.bambatch2-accepted_hits.bamchem1-accepted_hits.bamchem2-accepted_hits.bamDifferential_Counts_DESeq2_topTable_DESeq2.xls file. This file is a list of genes sorted by p-value from using DESeq2 to perform differential expression analysis.Differential_Counts_DESeq2.html file. This file has some output logs and plots from running DESeq2. Take a look at the PCA plot.PCA plots are useful for exploratory data analysis. Samples which are more similar to each other are expected to cluster together. A count matrix often has thousands of dimensions (one for each feature) and our PCA plot generated in the previous step transforms the data so the most variability is represented in principal components 1 and 2 (PC1 and PC2 represented by the x-axis and y-axis respectively).
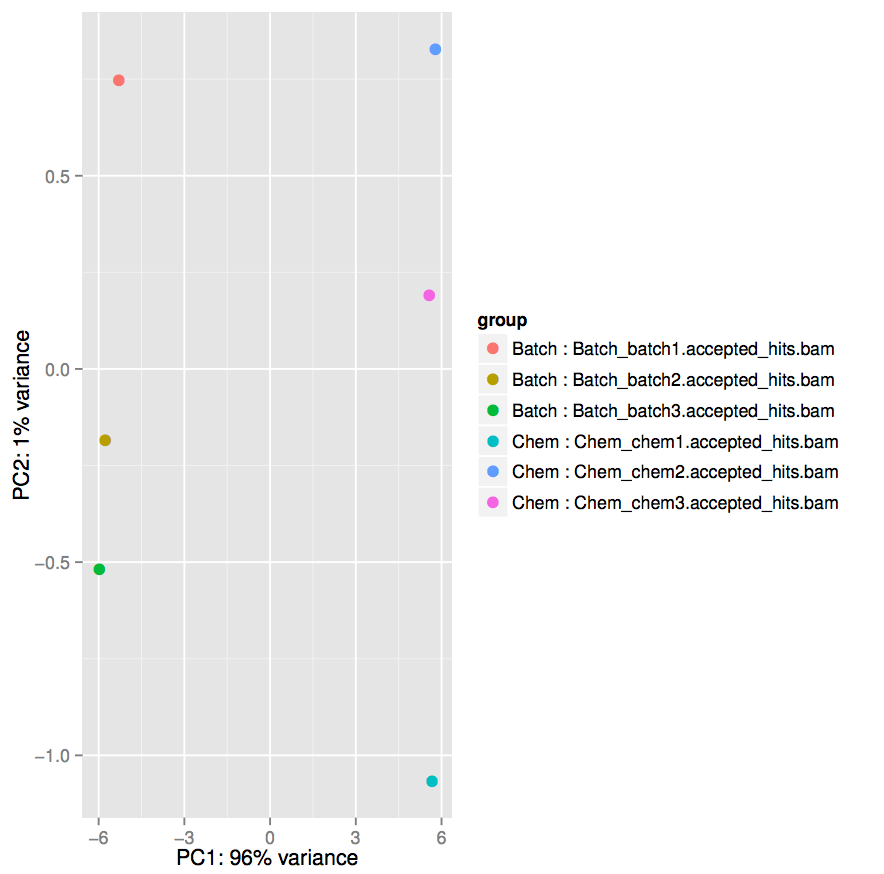
Take note of the scales on the x-axis and the y-axis. The x-axis representing the first principal component accounts for 96% of the variance and ranges from approximately -6 to +6, while the y-axis ranges from approximately -1 to +1.
For both conditions, the 3 replicates tend to be closer to each other than they are to replicates from the other condition.
Additionally, within conditions, the lower glucose (chem) condition shows more variability between replicates than the higher glucose (batch) condition.
Under Basic Tools, click on Filter and Sort > Filter:
Differential_Counts_DESeq2_topTable_DESeq2.xlsThis will keep the genes that have an adjusted p-value of less or equal to 0.05. There should be 53 genes in this file. Rename this file by clicking on the pencil icon of and change the name from "Filter on data x" to DESeq2_Significant_DE_Genes. You should see the first few differentially expressed genes are similar to the ones identified by EdgeR.
We are interested in how similar the identified genes are between the different statistial methods used by Cuffdiff, edgeR, and DESeq2. We can generate a Venn diagram to visualise the amount of overlap.
Cuffdiff_Significant_DE_GenesedgeR_Significant_DE_GenesDESeq2_Significant_DE_GenesView the generated Venn diagram. Agreement between the tools is good: there are 49 differentially expressed genes that all three tools agree upon, and only a handful that are exclusive to each tool.
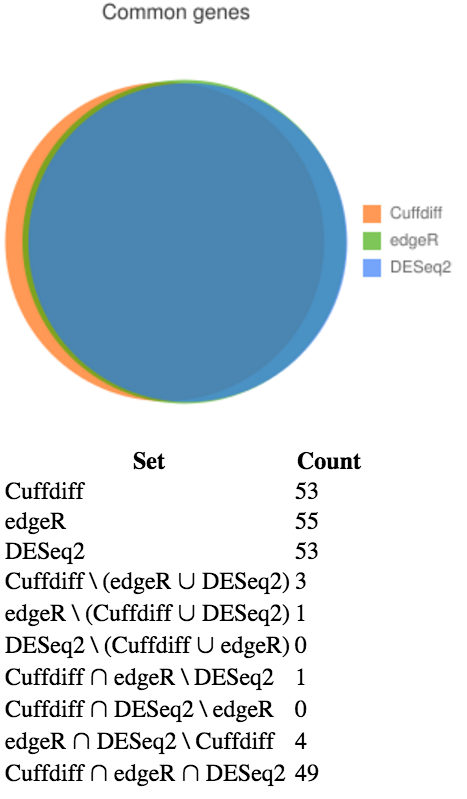
Generate the common list of significantly expressed genes identified by the three mentioned tools by extracting the respective gene list columns and intersecting:
Cuffdiff_Significant_DE_GenesRename the output to something like Cuffdiff_gene_list
edgeR_Significant_DE_GenesRename the output to something like edgeR_gene_list
DESeq2_Significant_DE_GenesRename the output to something like DESeq2_gene_list
Cuffdiff_gene_listedgeR_gene_listRename the output to something like Cuffdiff_edgeR_common_gene_list
Cuffdiff_edgeR_common_gene_listDESeq2_gene_listRename the output to something like Cuffdiff_edgeR_DESeq2_common_gene_list
We now have a list of 49 genes that have been identified as significantly differentially expressed by all three tools.
[1] Nookaew I, Papini M, Pornputtpong N, Scalcinati G, Fagerberg L, Uhlén M, Nielsen J: A comprehensive comparison of RNA-Seq-based transcriptome analysis from reads to differential gene expression and cross-comparison with microarrays: a case study in Saccharomyces cerevisiae. Nucleic Acids Res 2012, 40 (20):10084 – 10097. doi:10.1093/nar/gks804. Epub 2012 Sep 10
[2] Guirguis A, Slape C, Failla L, Saw J, Tremblay C, Powell D, Rossello F, Wei A, Strasser A, Curtis D: PUMA promotes apoptosis of hematopoietic progenitors driving leukemic progression in a mouse model of myelodysplasia. Cell Death Differ. 2016 Jun;23(6)