11 Cluster Markers
Why do we need to do this?
Single cell data helps to segragate cell types. Use markers to identify cell types. warning: In this example the cell types/markers are well known and making this step easy, but in reality this step needs the experts curation.
11.1 Finding differentially expressed features (cluster biomarkers)
Seurat can help you find markers that define clusters via differential expression. By default, it identifies positive and negative markers of a single cluster (specified in ident.1), compared to all other cells. FindAllMarkers() automates this process for all clusters, but you can also test groups of clusters vs. each other, or against all cells.
The min.pct argument requires a feature to be detected at a minimum percentage in either of the two groups of cells, and the thresh.test argument requires a feature to be differentially expressed (on average) by some amount between the two groups. You can set both of these to 0, but with a dramatic increase in time - since this will test a large number of features that are unlikely to be highly discriminatory. As another option to speed up these computations, max.cells.per.ident can be set. This will downsample each identity class to have no more cells than whatever this is set to. While there is generally going to be a loss in power, the speed increases can be significant and the most highly differentially expressed features will likely still rise to the top.
# find all markers of cluster 2
cluster2.markers <- FindMarkers(seurat_object, ident.1 = 2, min.pct = 0.25)
head(cluster2.markers, n = 5)
#> p_val avg_log2FC pct.1 pct.2 p_val_adj
#> NKG7 0 5.661923 0.800 0.056 0
#> CCL5 0 4.482005 0.749 0.107 0
#> GZMB 0 5.281761 0.652 0.037 0
#> GNLY 0 6.095800 0.661 0.049 0
#> CST7 0 4.799111 0.627 0.044 0
# find all markers distinguishing cluster 5 from clusters 0 and 3
cluster5.markers <- FindMarkers(seurat_object, ident.1 = 5, ident.2 = c(0, 3), min.pct = 0.25)
head(cluster5.markers, n = 5)
#> p_val avg_log2FC pct.1 pct.2 p_val_adj
#> HSPH1 2.191916e-116 4.608521 0.591 0.060 7.810891e-112
#> KLF10 7.713460e-63 4.173941 0.274 0.020 2.748692e-58
#> HSPA1A 1.488828e-62 5.250111 0.378 0.046 5.305439e-58
#> SRSF2 1.282249e-60 2.836025 0.689 0.184 4.569294e-56
#> SNHG15 9.077911e-60 3.667237 0.390 0.051 3.234914e-55
# find markers for every cluster compared to all remaining cells, report only the positive ones
seurat_object.markers <- FindAllMarkers(seurat_object, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
#> Calculating cluster 0
#> Calculating cluster 1
#> Calculating cluster 2
#> Calculating cluster 3
#> Calculating cluster 4
#> Calculating cluster 5
#> Calculating cluster 6
#> Calculating cluster 7
seurat_object.markers %>% group_by(cluster) %>% slice_max(n = 2, order_by = avg_log2FC)
#> # A tibble: 16 × 7
#> # Groups: cluster [8]
#> p_val avg_log2FC pct.1 pct.2 p_val_adj cluster gene
#> <dbl> <dbl> <dbl> <dbl> <dbl> <fct> <chr>
#> 1 8.51e-196 2.11 0.614 0.223 3.03e-191 0 SELL
#> 2 3.26e-223 2.05 0.653 0.221 1.16e-218 0 LTB
#> 3 0 7.02 0.672 0.021 0 1 S100A8
#> 4 2.19e-267 7.00 0.353 0.013 7.80e-263 1 CCL7
#> 5 0 6.10 0.661 0.049 0 2 GNLY
#> 6 4.38e-255 6.07 0.331 0.011 1.56e-250 2 GZMA
#> 7 0 6.29 0.442 0.011 0 3 MS4A1
#> 8 0 6.17 0.657 0.018 0 3 CD79A
#> 9 0 6.75 0.833 0.029 0 4 VMO1
#> 10 0 5.19 0.742 0.036 0 4 MS4A4A
#> 11 2.18e-121 4.63 0.591 0.076 7.76e-117 5 HSPH1
#> 12 3.46e- 90 4.21 0.39 0.04 1.23e- 85 5 SNHG15
#> 13 2.72e-243 7.17 0.438 0.011 9.68e-239 6 PF4
#> 14 1.51e-120 6.57 0.253 0.009 5.38e-116 6 SDPR
#> 15 0 8.79 0.438 0.001 0 7 CCL22
#> 16 0 7.42 0.517 0.003 0 7 PKIBSeurat has several tests for differential expression which can be set with the test.use parameter (see our DE vignette for details). For example, the ROC test returns the ‘classification power’ abs(AUC-0.5)*2 for any individual marker, ranging from 0 = random to 1 = perfect.
cluster0.markers <- FindMarkers(seurat_object, ident.1 = 0, logfc.threshold = 0.25, test.use = "roc", only.pos = TRUE)We include several tools for visualizing marker expression. VlnPlot() (shows expression probability distributions across clusters), and FeaturePlot() (visualizes feature expression on a tSNE or PCA plot) are our most commonly used visualizations. We also suggest exploring RidgePlot(), CellScatter(), and DotPlot() as additional methods to view your dataset.
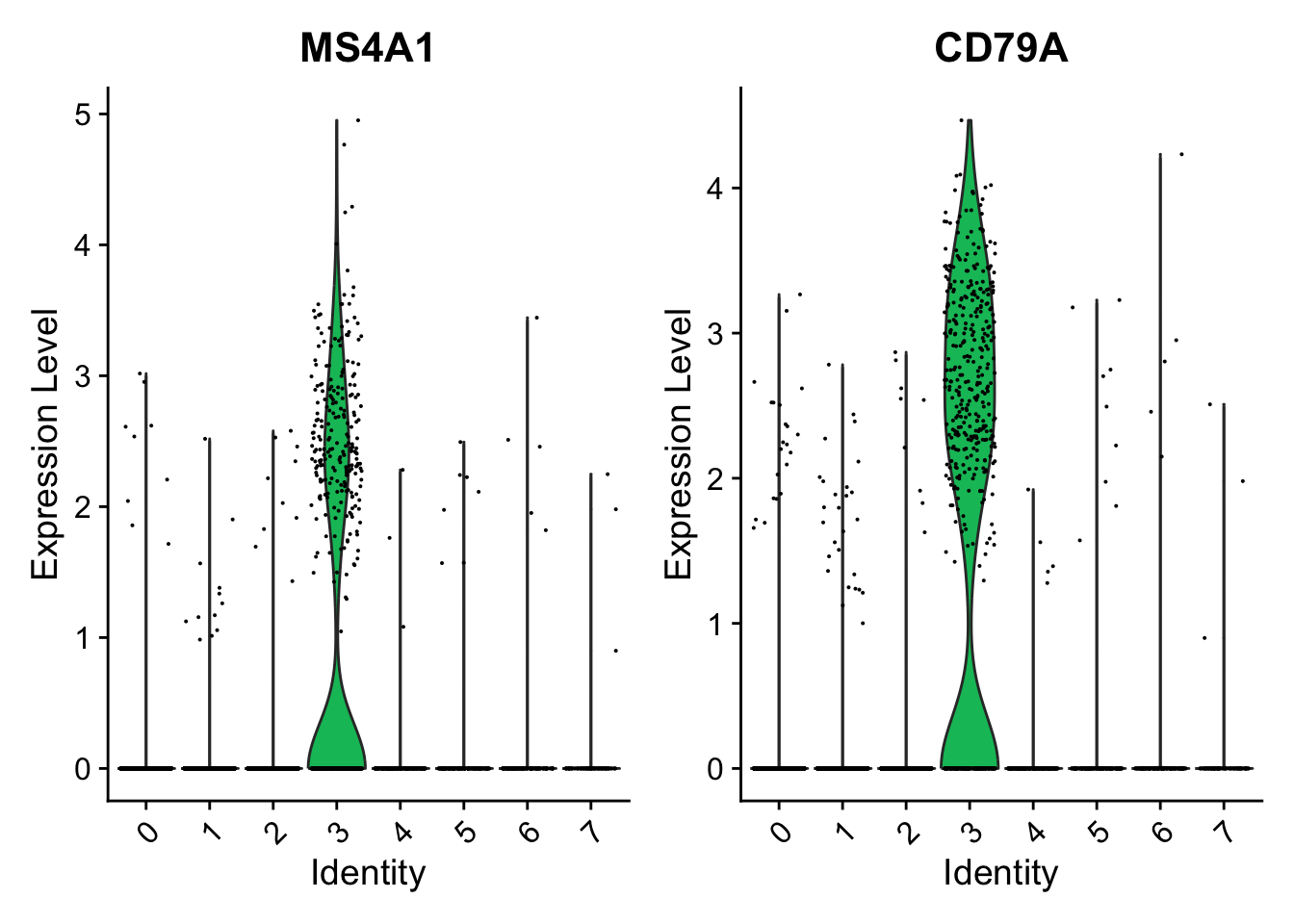
# you can plot raw counts as well
VlnPlot(seurat_object, features = c("NKG7", "PF4"), slot = 'counts', log = TRUE)
#> Warning: The `slot` argument of `VlnPlot()` is deprecated as of Seurat 5.0.0.
#> ℹ Please use the `layer` argument instead.
#> This warning is displayed once every 8 hours.
#> Call `lifecycle::last_lifecycle_warnings()` to see where this warning was generated.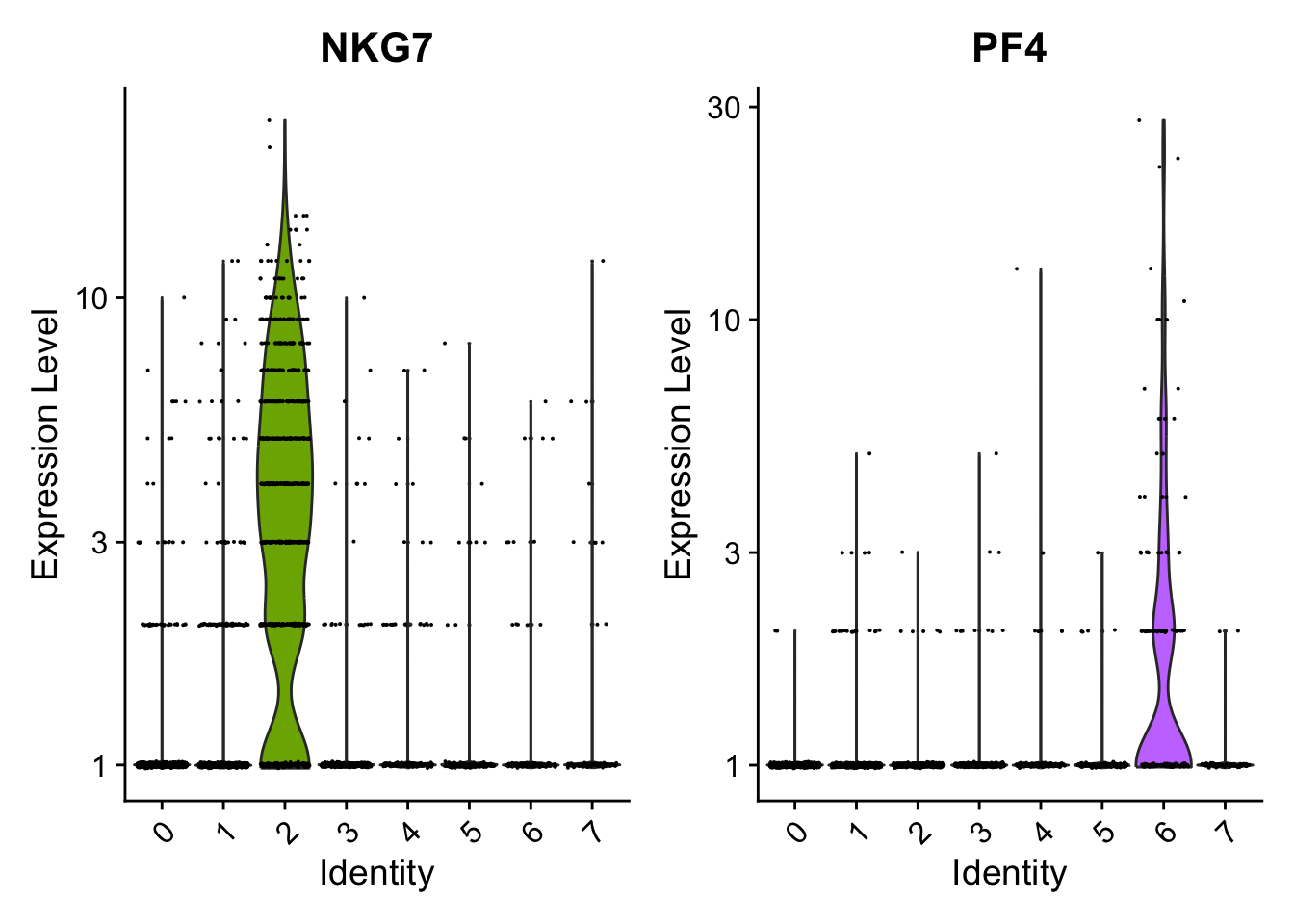
FeaturePlot(seurat_object, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A"))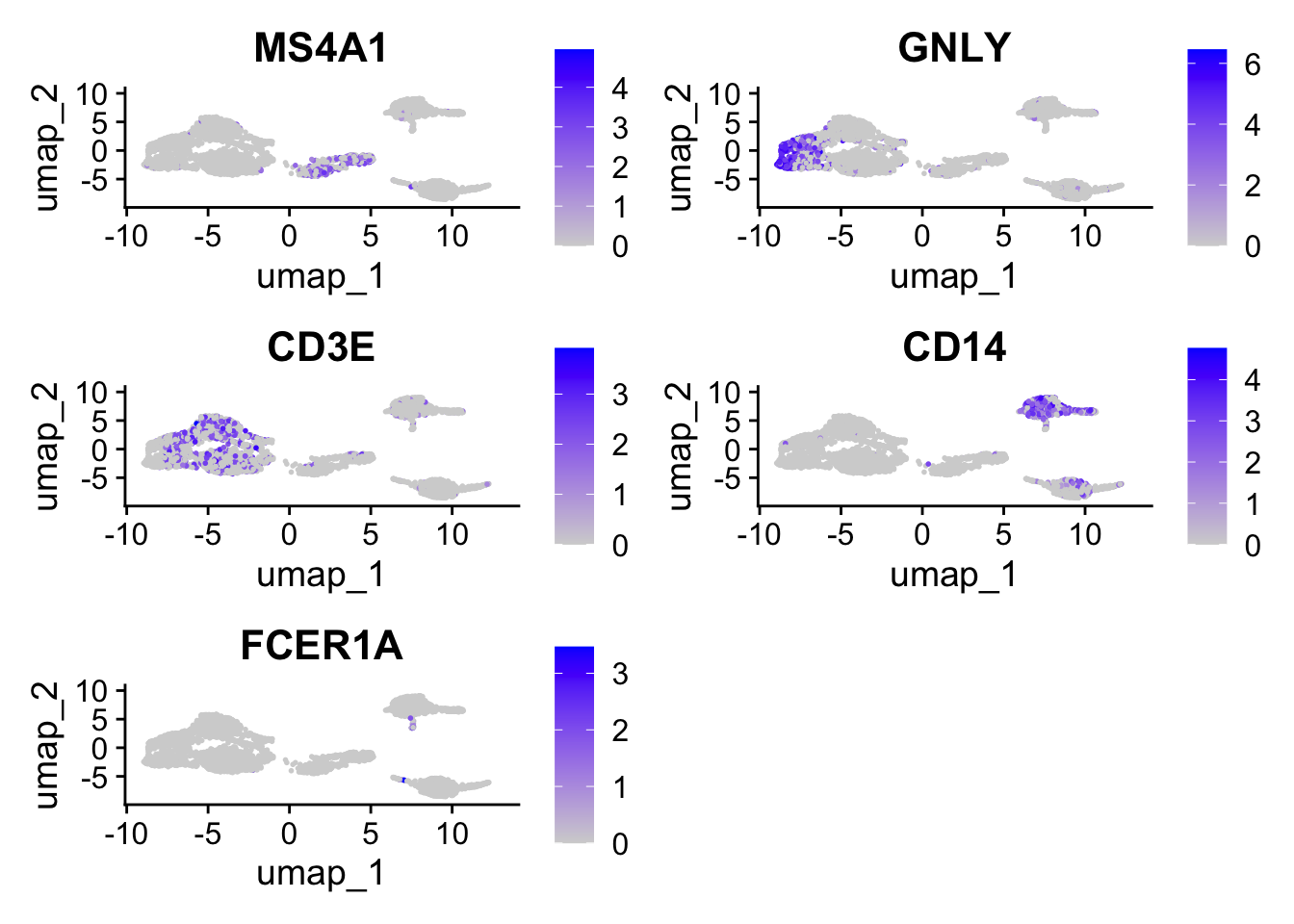
FeaturePlot(seurat_object, features = c("FCGR3A", "LYZ", "PPBP", "CD8A"))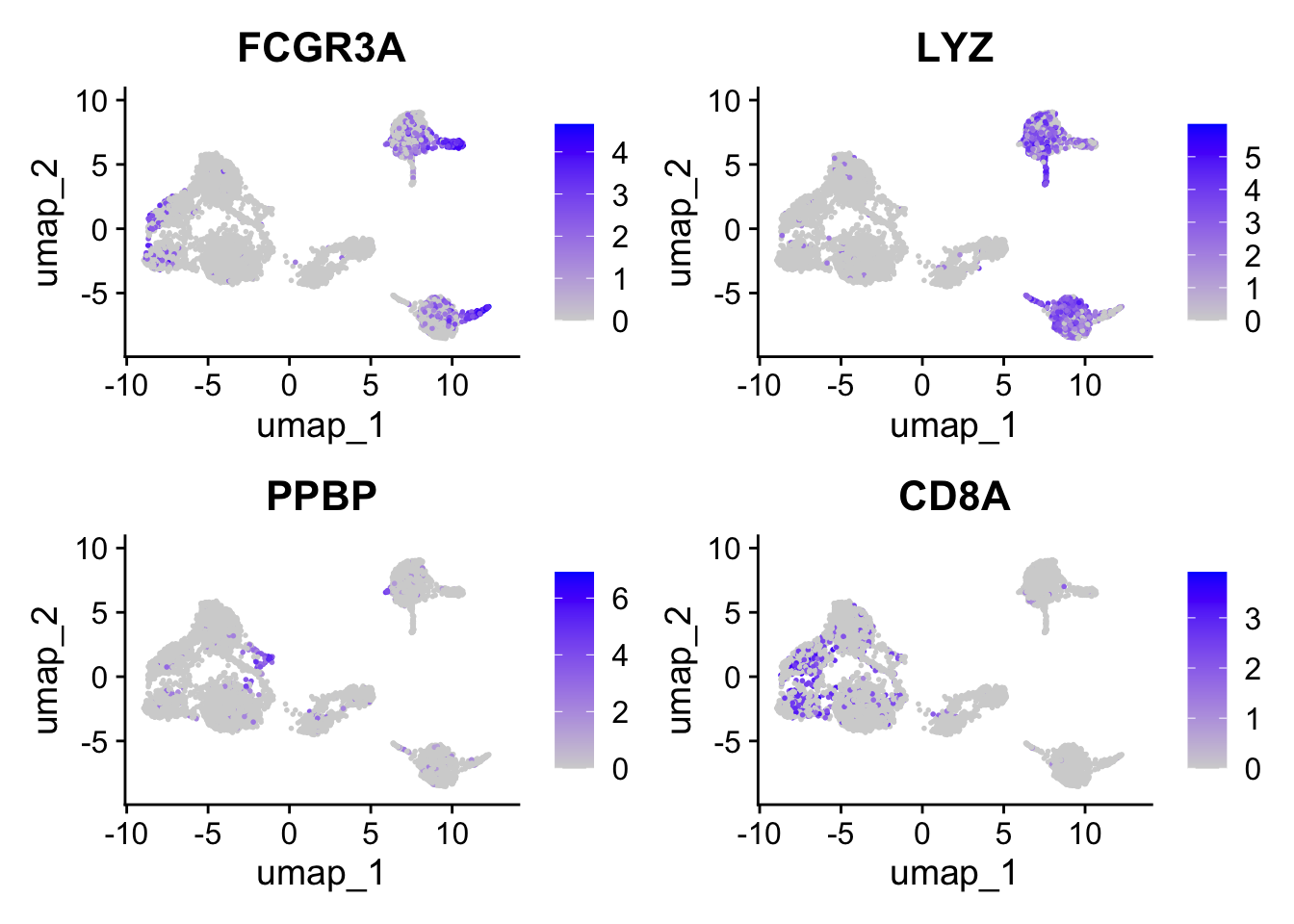
Other useful plots
These are ridgeplots, cell scatter plots and dotplots. Replace FeaturePlot with the other functions.
RidgePlot(seurat_object, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A"))
#> Picking joint bandwidth of 0.0426
#> Picking joint bandwidth of 0.0658
#> Picking joint bandwidth of 0.0723
#> Picking joint bandwidth of 0.0901
#> Picking joint bandwidth of 2.99e-06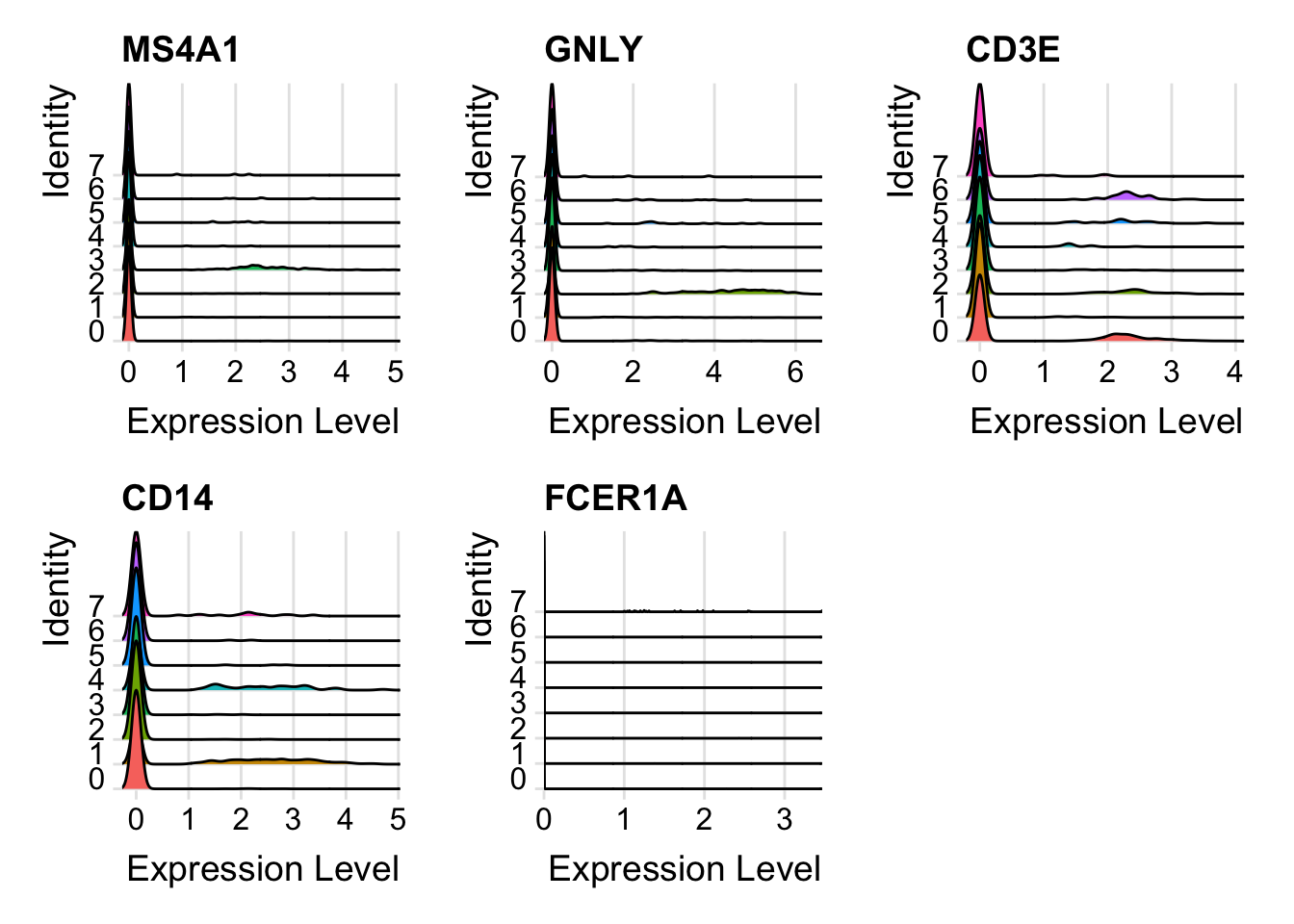
RidgePlot(seurat_object, features = c("FCGR3A", "LYZ", "PPBP", "CD8A"))
#> Picking joint bandwidth of 0.0546
#> Picking joint bandwidth of 0.136
#> Picking joint bandwidth of 0.0843
#> Picking joint bandwidth of 0.0343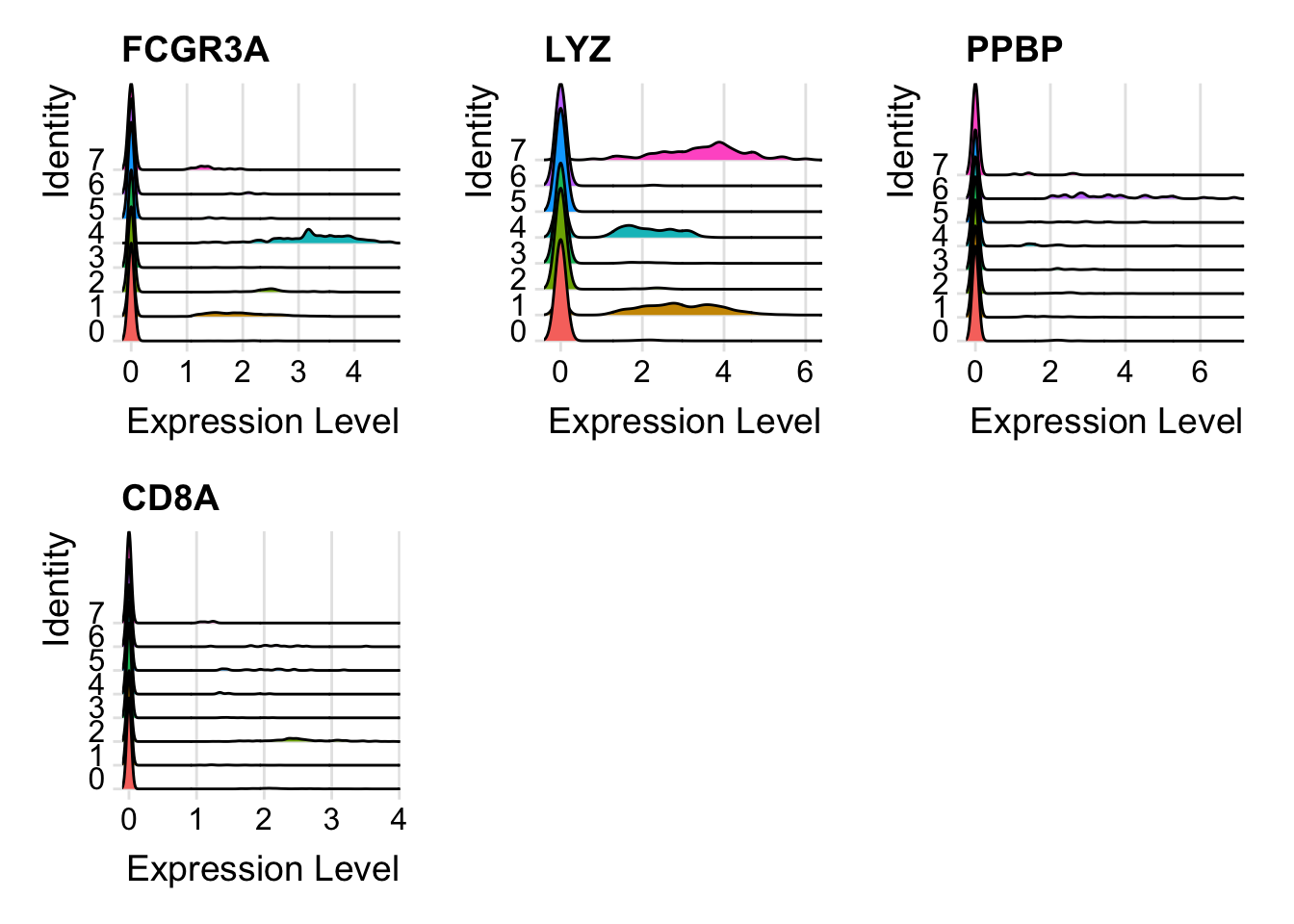 For CellScatter plots, will need the cell id of the cells you want to look at. You can get this from the cell metadata (
For CellScatter plots, will need the cell id of the cells you want to look at. You can get this from the cell metadata (seurat_object@meta.data).
head( seurat_object@meta.data )
#> orig.ident nCount_RNA nFeature_RNA ind
#> AGGGCGCTATTTCC-1 SeuratProject 2053 532 1256
#> GGAGACGATTCGTT-1 SeuratProject 881 392 1256
#> CACCGTTGTCGTAG-1 SeuratProject 3130 1005 1016
#> TATCGTACACGCAT-1 SeuratProject 1042 549 1488
#> TACGAGACCTATTC-1 SeuratProject 2425 777 1244
#> GTACTACTCATACG-1 SeuratProject 3951 1064 1256
#> stim cell multiplets
#> AGGGCGCTATTTCC-1 stim CD14+ Monocytes singlet
#> GGAGACGATTCGTT-1 stim CD4 T cells singlet
#> CACCGTTGTCGTAG-1 ctrl FCGR3A+ Monocytes singlet
#> TATCGTACACGCAT-1 stim B cells singlet
#> TACGAGACCTATTC-1 stim CD4 T cells singlet
#> GTACTACTCATACG-1 ctrl FCGR3A+ Monocytes singlet
#> percent.mt RNA_snn_res.0.5 seurat_clusters
#> AGGGCGCTATTTCC-1 1.6336634 1 0
#> GGAGACGATTCGTT-1 4.8809524 6 15
#> CACCGTTGTCGTAG-1 1.0655473 4 11
#> TATCGTACACGCAT-1 3.0662710 3 4
#> TACGAGACCTATTC-1 1.0837849 0 8
#> GTACTACTCATACG-1 0.7137395 4 11
#> pca_clusters harmony_clusters
#> AGGGCGCTATTTCC-1 3 1
#> GGAGACGATTCGTT-1 0 6
#> CACCGTTGTCGTAG-1 9 4
#> TATCGTACACGCAT-1 6 3
#> TACGAGACCTATTC-1 0 0
#> GTACTACTCATACG-1 9 4
#> RNA_snn_res.0.1 RNA_snn_res.0.2
#> AGGGCGCTATTTCC-1 1 1
#> GGAGACGATTCGTT-1 0 0
#> CACCGTTGTCGTAG-1 4 4
#> TATCGTACACGCAT-1 3 3
#> TACGAGACCTATTC-1 0 0
#> GTACTACTCATACG-1 4 4
#> RNA_snn_res.0.3 RNA_snn_res.0.4
#> AGGGCGCTATTTCC-1 1 1
#> GGAGACGATTCGTT-1 0 6
#> CACCGTTGTCGTAG-1 4 4
#> TATCGTACACGCAT-1 3 3
#> TACGAGACCTATTC-1 0 0
#> GTACTACTCATACG-1 4 4
#> RNA_snn_res.0.6 RNA_snn_res.0.7
#> AGGGCGCTATTTCC-1 1 1
#> GGAGACGATTCGTT-1 7 7
#> CACCGTTGTCGTAG-1 5 6
#> TATCGTACACGCAT-1 3 4
#> TACGAGACCTATTC-1 0 3
#> GTACTACTCATACG-1 5 6
#> RNA_snn_res.0.8 RNA_snn_res.0.9
#> AGGGCGCTATTTCC-1 1 1
#> GGAGACGATTCGTT-1 9 9
#> CACCGTTGTCGTAG-1 7 7
#> TATCGTACACGCAT-1 3 3
#> TACGAGACCTATTC-1 0 0
#> GTACTACTCATACG-1 7 7
#> RNA_snn_res.1 RNA_snn_res.1.1
#> AGGGCGCTATTTCC-1 0 1
#> GGAGACGATTCGTT-1 9 8
#> CACCGTTGTCGTAG-1 8 7
#> TATCGTACACGCAT-1 3 4
#> TACGAGACCTATTC-1 6 0
#> GTACTACTCATACG-1 8 7
#> RNA_snn_res.1.2 RNA_snn_res.1.3
#> AGGGCGCTATTTCC-1 0 2
#> GGAGACGATTCGTT-1 10 11
#> CACCGTTGTCGTAG-1 8 9
#> TATCGTACACGCAT-1 3 3
#> TACGAGACCTATTC-1 2 7
#> GTACTACTCATACG-1 8 9
#> RNA_snn_res.1.4 RNA_snn_res.1.5
#> AGGGCGCTATTTCC-1 0 1
#> GGAGACGATTCGTT-1 12 12
#> CACCGTTGTCGTAG-1 10 10
#> TATCGTACACGCAT-1 1 3
#> TACGAGACCTATTC-1 2 4
#> GTACTACTCATACG-1 10 10
#> RNA_snn_res.1.6 RNA_snn_res.1.7
#> AGGGCGCTATTTCC-1 1 0
#> GGAGACGATTCGTT-1 12 13
#> CACCGTTGTCGTAG-1 10 11
#> TATCGTACACGCAT-1 2 1
#> TACGAGACCTATTC-1 9 7
#> GTACTACTCATACG-1 10 11
#> RNA_snn_res.1.8 RNA_snn_res.1.9
#> AGGGCGCTATTTCC-1 0 7
#> GGAGACGATTCGTT-1 13 14
#> CACCGTTGTCGTAG-1 11 12
#> TATCGTACACGCAT-1 1 0
#> TACGAGACCTATTC-1 8 8
#> GTACTACTCATACG-1 11 12
#> RNA_snn_res.2
#> AGGGCGCTATTTCC-1 0
#> GGAGACGATTCGTT-1 15
#> CACCGTTGTCGTAG-1 11
#> TATCGTACACGCAT-1 4
#> TACGAGACCTATTC-1 8
#> GTACTACTCATACG-1 11
CellScatter(seurat_object, cell1 = "AGGGCGCTATTTCC-1", cell2 = "GGAGACGATTCGTT-1")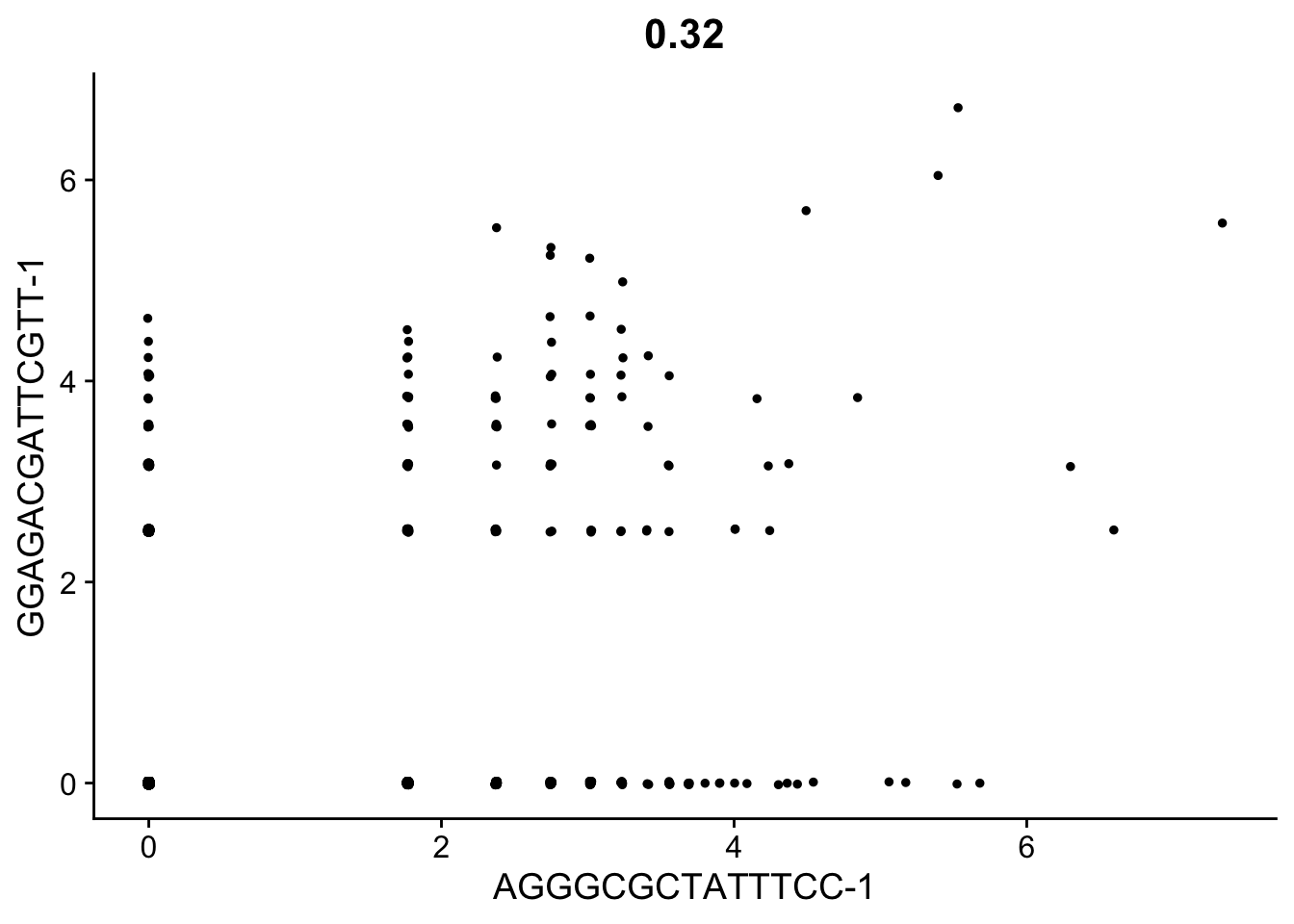
CellScatter(seurat_object, cell1 = "GGAGACGATTCGTT-1", cell2 = "TACGAGACCTATTC-1")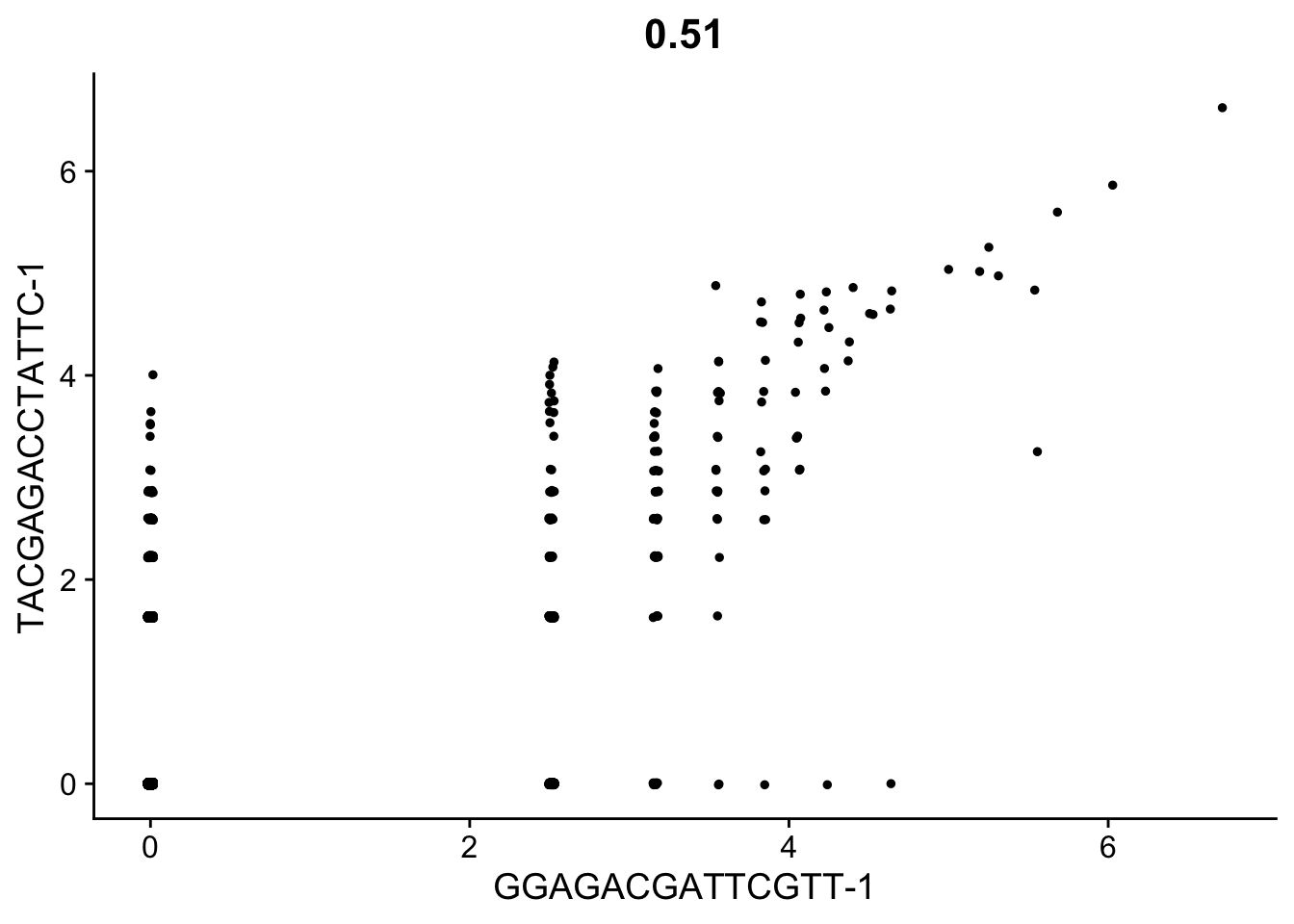
DotPlots
DotPlot(seurat_object, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP", "CD8A"))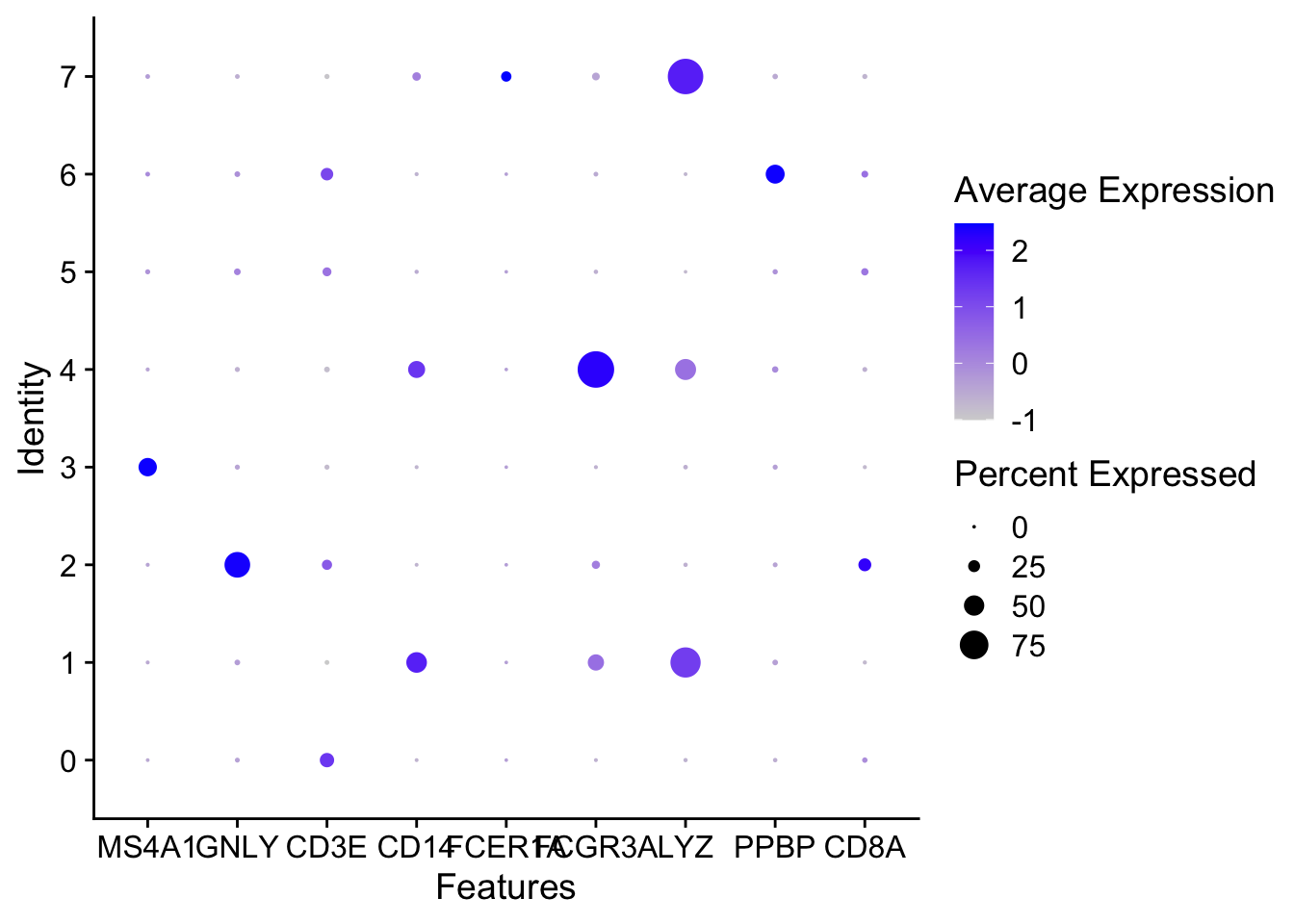
DoHeatmap() generates an expression heatmap for given cells and features. In this case, we are plotting the top 10 markers (or all markers if less than 10) for each cluster.
maxcells=1500
top10 <- seurat_object.markers %>% group_by(cluster) %>% top_n(n = 10, wt = avg_log2FC)
DoHeatmap(subset(seurat_object, downsample = maxcells), features = top10$gene) + NoLegend()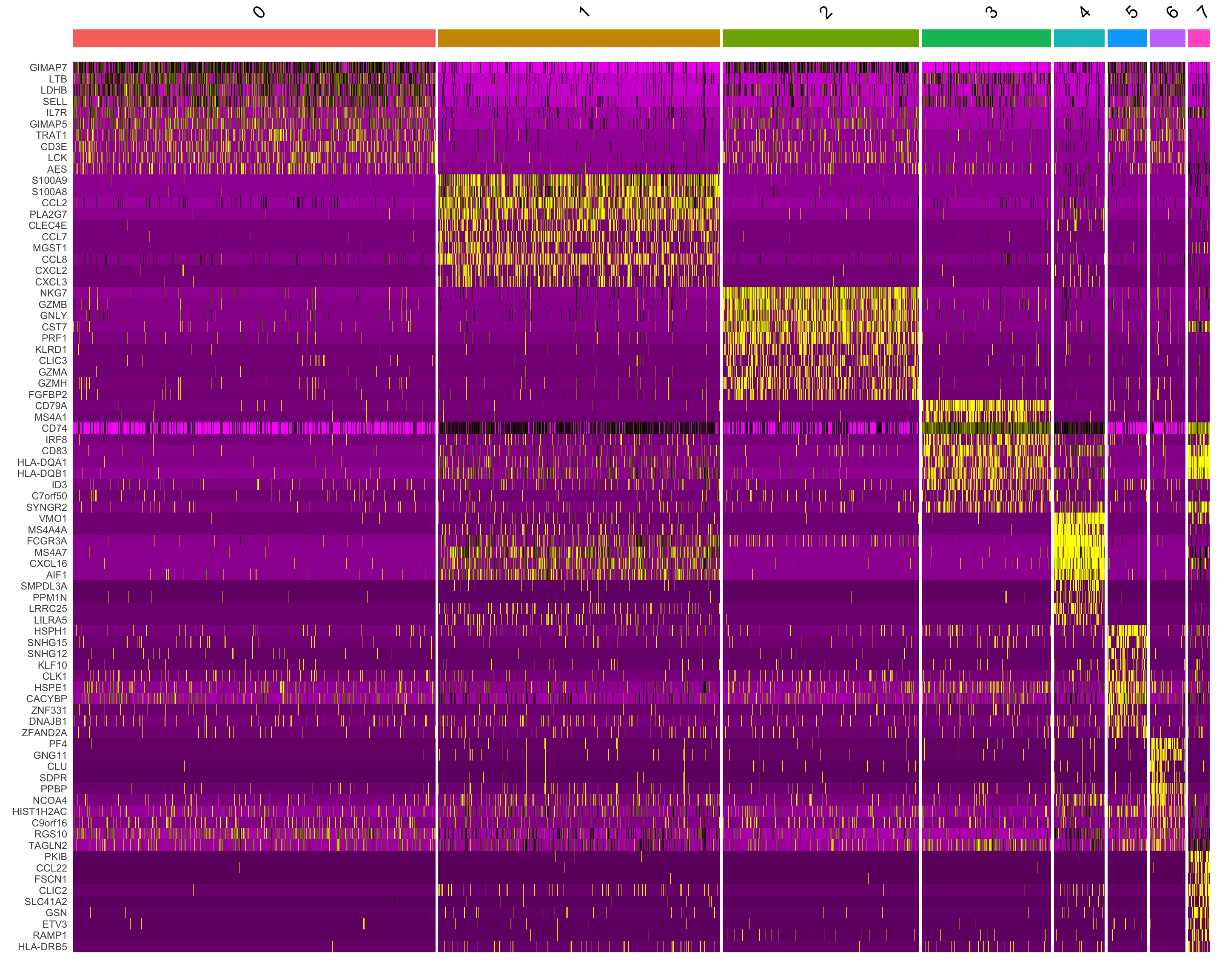
11.2 Use makers to label or find a cluster
If you know markers for your cell types, use AddModuleScore to label them.
genes_markers <- list(Naive_CD4_T = c("IL7R", "CCR7"))
seurat_object <- AddModuleScore(object = seurat_object, features = genes_markers, ctrl = 5, name = "Naive_CD4_T",
search = TRUE)
# notice the name of the cluster has a 1 at the end
names(seurat_object@meta.data)
#> [1] "orig.ident" "nCount_RNA"
#> [3] "nFeature_RNA" "ind"
#> [5] "stim" "cell"
#> [7] "multiplets" "percent.mt"
#> [9] "RNA_snn_res.0.5" "seurat_clusters"
#> [11] "pca_clusters" "harmony_clusters"
#> [13] "RNA_snn_res.0.1" "RNA_snn_res.0.2"
#> [15] "RNA_snn_res.0.3" "RNA_snn_res.0.4"
#> [17] "RNA_snn_res.0.6" "RNA_snn_res.0.7"
#> [19] "RNA_snn_res.0.8" "RNA_snn_res.0.9"
#> [21] "RNA_snn_res.1" "RNA_snn_res.1.1"
#> [23] "RNA_snn_res.1.2" "RNA_snn_res.1.3"
#> [25] "RNA_snn_res.1.4" "RNA_snn_res.1.5"
#> [27] "RNA_snn_res.1.6" "RNA_snn_res.1.7"
#> [29] "RNA_snn_res.1.8" "RNA_snn_res.1.9"
#> [31] "RNA_snn_res.2" "Naive_CD4_T1"
# label that cell type
seurat_object$cell_label = NA
seurat_object$cell_label[seurat_object$Naive_CD4_T1 > 1] = "Naive_CD4_T"
Idents(seurat_object) = seurat_object$cell_label
# plot
# Using a custom colour scale
FeaturePlot(seurat_object, features = "Naive_CD4_T1", label = TRUE, repel = TRUE, reduction = "umap_harmony") + scale_colour_gradientn(colours = c("lightblue","beige","red"))
#> Scale for colour is already present.
#> Adding another scale for colour, which will replace the existing scale.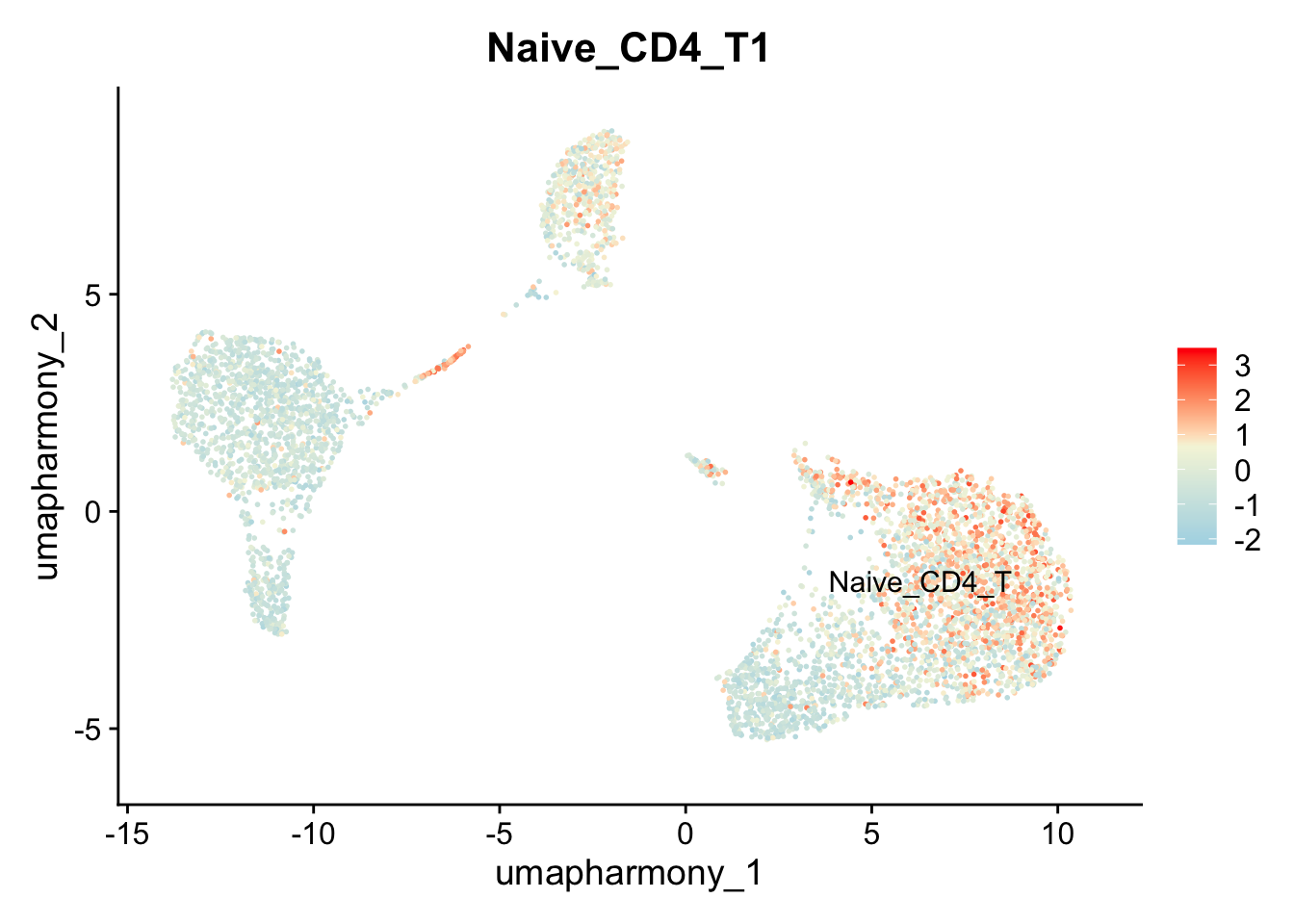
11.3 Assigning cell type identity to clusters
Fortunately in the case of this dataset, we can use canonical markers to easily match the unbiased clustering to known cell types:
| Markers | Cell Type |
|---|---|
| IL7R, CCR7 | Naive CD4+ T |
| CD14, LYZ | CD14+ Mono |
| IL7R, S100A4 | Memory CD4+ |
| MS4A1 | B |
| CD8A | CD8+ T |
| FCGR3A, MS4A7 | FCGR3A+ Mono |
| GNLY, NKG7 | NK |
| FCER1A, CST3 | DC |
| PPBP | Platelet |
DimPlot(seurat_object,group.by = "RNA_snn_res.0.5",reduction = "umap_harmony")|FeaturePlot(seurat_object, features = c( "MS4A1"),reduction = "umap_harmony")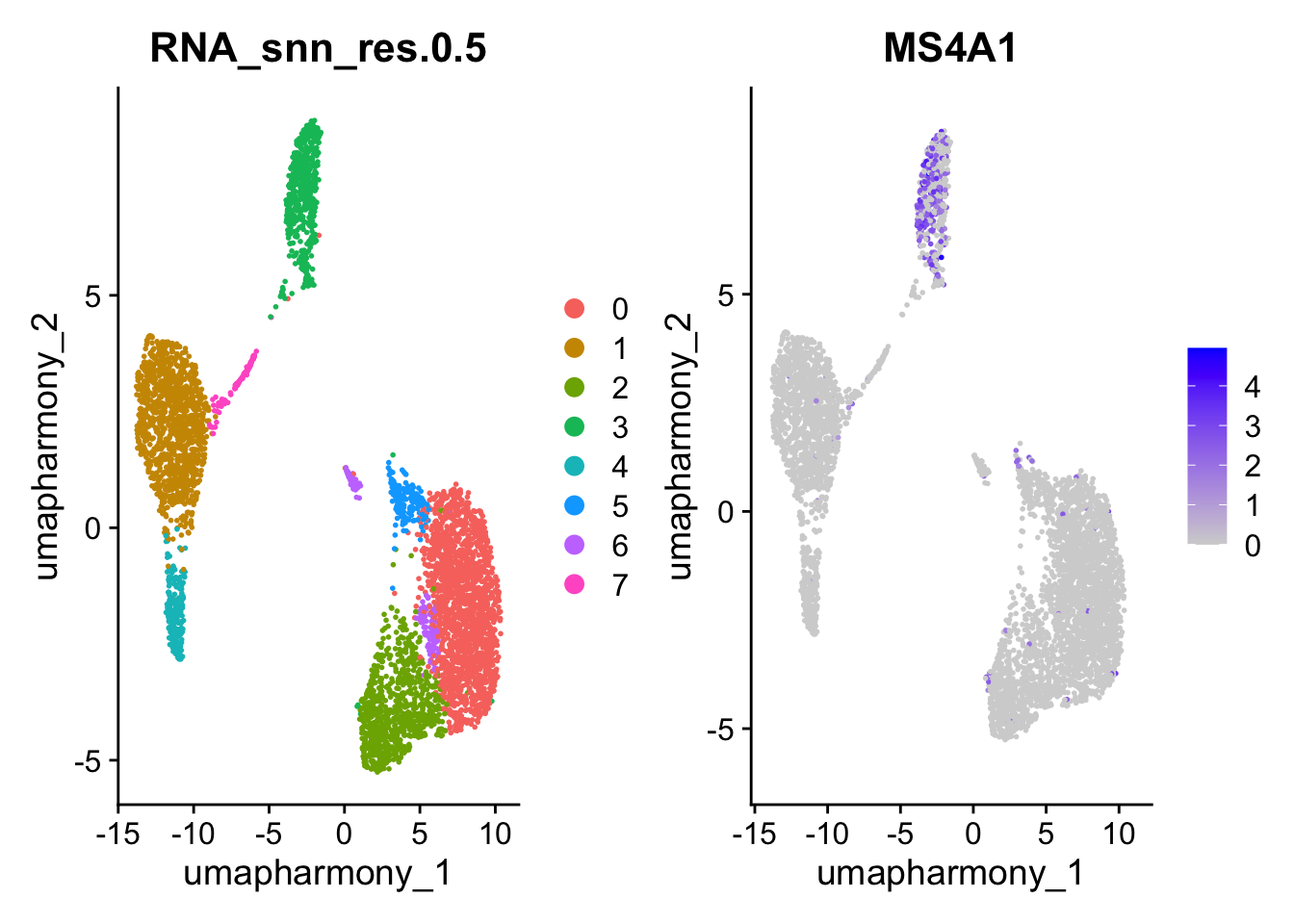
Challenge: Match cluster numbers with cell labels
Use the markers provided and the resolution 0.5 to identity the cell labels
Idents(seurat_object) <- seurat_object$RNA_snn_res.0.5
# This renaming list is incomplete and incorrect.
# Fix and add to the list to fill in the cell types.
new.cluster.ids <- c(
"0" = "Naive CD4+ T",
"2" = "CD14+ Monocytes",
"1" = "B cells")
seurat_object <- RenameIdents(seurat_object, new.cluster.ids)
DimPlot(seurat_object, reduction = 'umap_harmony', label = TRUE, pt.size = 0.5) + NoLegend()11.3.0.1 save the plots
plot <- DimPlot(seurat_object, reduction = 'umap_harmony', label = TRUE, label.size = 4.5) + xlab("UMAP 1") + ylab("UMAP 2") +
theme(axis.title = element_text(size = 18), legend.text = element_text(size = 18)) +
guides(colour = guide_legend(override.aes = list(size = 10)))
ggsave(filename = "seurat_object3k_umap.jpg", height = 7, width = 12, plot = plot, quality = 50)11.3.0.2 save the seurat object
saveRDS(seurat_object, file = "seurat_object3k_final.rds")